
Pharmaceutical innovation stems from a variety of sources. Practicing doctors and healthcare providers generate concepts and collect observational data for new and improved treatments using existing approved medication. Alternatively, university researchers study existing molecules for different disease models in vitro or in vivo. Pharmaceutical companies license these assets and undertake preclinical and clinical development with the goal of filing an NDA for FDA approval. Companies need a clear understanding of the available FDA approval pathways prior to undertaking preclinical and clinical development. There are three available FDA approval pathways for pharmaceutical products:
- 505(b)(1)
- 505(b)(2)
- 505(j)
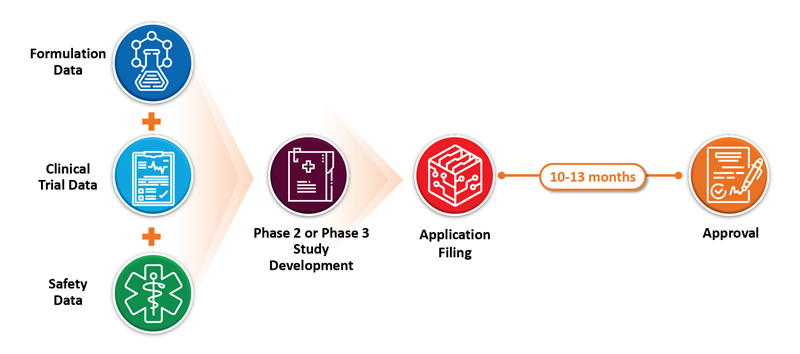 A 505(b)(1) application must contain full reports of investigations of the safety and effectiveness of the drug. This is typically needed for new chemical entities (NCEs) and requires sponsors to perform pre-clinical studies, clinical studies, and development on the drug substance and drug product’s chemistry, manufacturing, and controls (CMC). Such programs tend to be expensive and require a long period to complete. On the other extreme, 505(j) applications require that an applicant demonstrate that the proposed product is identical in active pharmaceutical ingredient (API), route of administration, dosage form, strength, labeling, quality, performance characteristics, and intended use when compared to an approved reference listed drug (RLD). These are referred to as ANDA (abbreviated new drug application) filings. These programs typically cost much less than NDA programs and require 2 to 4 years from start to launch. Post-approval clinical studies are not required, and sales and marketing costs are minimal.
A 505(b)(1) application must contain full reports of investigations of the safety and effectiveness of the drug. This is typically needed for new chemical entities (NCEs) and requires sponsors to perform pre-clinical studies, clinical studies, and development on the drug substance and drug product’s chemistry, manufacturing, and controls (CMC). Such programs tend to be expensive and require a long period to complete. On the other extreme, 505(j) applications require that an applicant demonstrate that the proposed product is identical in active pharmaceutical ingredient (API), route of administration, dosage form, strength, labeling, quality, performance characteristics, and intended use when compared to an approved reference listed drug (RLD). These are referred to as ANDA (abbreviated new drug application) filings. These programs typically cost much less than NDA programs and require 2 to 4 years from start to launch. Post-approval clinical studies are not required, and sales and marketing costs are minimal.
The 505(b)(2) regulatory pathway falls between the other two pathways in almost every sense: development cost, complexity, and time.
- The product must be identical in active ingredient (API) to a previously approved reference product.
- A sponsor is not eligible for a 505(b)(2) application if the product fits the criteria of a 505(j) filing. There must be substantial change from previously approved products.
- A sponsor must submit a 505(b)(2) application if the sponsor must rely on published literature. However, there are specific requirements for such literature to be acceptable to the FDA.
- A 505(b)(2) application may be submitted for an NCE when some part of the data necessary for approval is derived from studies not conducted by or for the applicant and to which the applicant has not obtained a right of reference.
- Since pharmacy substation is not possible, sponsors must undertake their own sales and marketing efforts. Also, 505(b)(2) filings are subject to post-approval clinical requirements.
Typical reasons for selecting the 505(b)(2) filings pathway include changes to strength, indication, route of administration, dosage form, or frequency of administration. Many 505(b)(2) products have been approved without the need for animal studies, safety studies, or even efficacy studies. The requirements depend on the nature of the intended product, available literature data, and the reference product. A properly executed pre-IND communication strategy and the selection of an experienced CMC and formulation development team will save the sponsor time and money.
Vici Health Sciences has considerable experience in various aspects of 505(b)(2) development programs. This includes formulation and regulatory strategy, writing pre-IND and IND sections, designing and developing novel patent-protected formulations, creating IP, assisting with nonclinical studies when needed, manufacturing first-in-human phase 1 and phase 2 cGMP clinical supplies, project managing all aspects of R&D, and managing CMO tech transfer for phase 3 supplies manufacturing and NDA 505(b)(2) filings. Reach out to us for a free consultation in your program.
To contact our team, please email info@vicihealthsciences.com, or call +1 (410) 379-1500.