


Quality and safety are of utmost importance, among other critical factors, when developing a new drug. But clinical and analytical departments sometimes struggle to decide at which phase cGMP should be incorporated.
What is cGMP?
The acronym cGMP stands for Current Good Manufacturing Practices. These are regulations that are enforced by the FDA to ensure that manufacturing systems are designed and monitored to meet quality standards during drug development. The “current” in cGMP indicates that the processes are continuously improved with updated guidelines.
What is the role of cGMP in drug development?
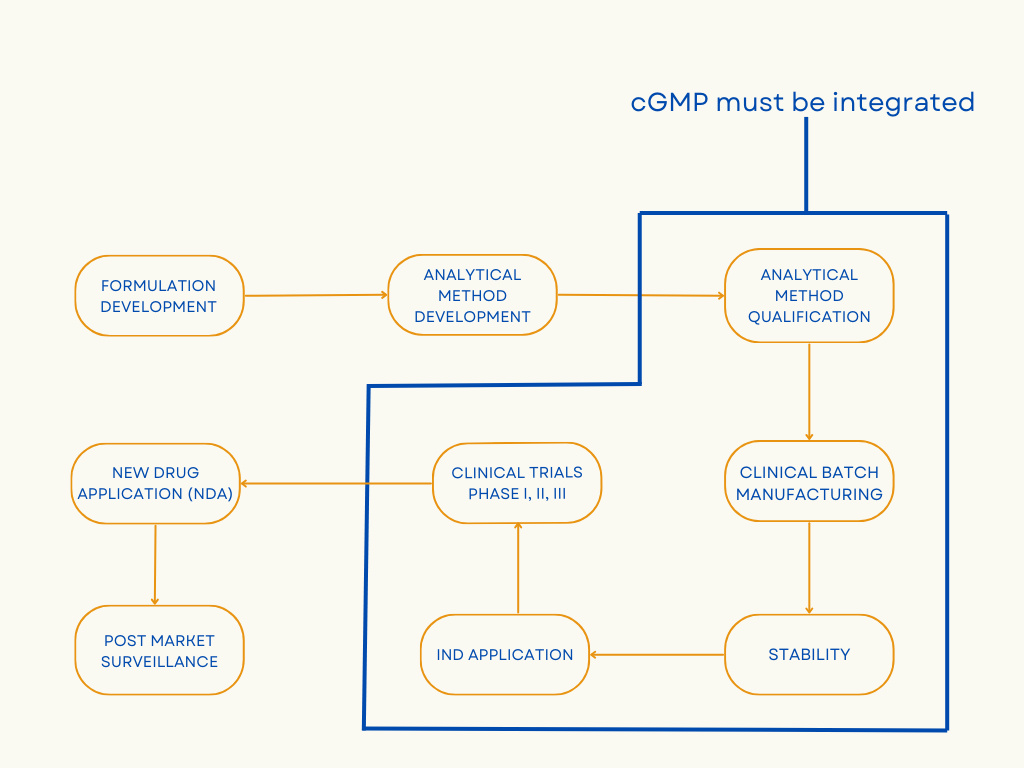
Initial stages of drug development are typically not performed in labs operating under cGMP. Early phase work is exploratory in nature and data from this stage may not be used to support human clinical trials or even be part of IND or NDA filings. As program enters the clinical phase of drug development, patient safety and compliance with FDA regulations are very important.
-
- Analytical method qualification/validation: During method validation, the data collected during method development is evaluated to conclude that the process can consistently deliver quality products. Method qualification is conducted to evaluate the process performance and determine if the process can deliver reproducible batches for commercialization. Method validation and qualification streamlines the manufacturing process and allows for a smooth transition from clinical to commercialization.
-
- Raw materials utilized in manufacturing clinical supplies: Vendor qualification is performed by auditing vendor sites and verifying the raw material specifications mentioned on the vendor CoA. Vendors are given cGMP certification based on but not limited to the maintenance of storage conditions for raw materials, design and cleanliness maintained at the facility, qualification of ingredients and labels.
-
- Clinical batch manufacturing: When clinical batches are manufactured, the right combination of equipment qualification, validation, and comprehensive documentation is required for cGMP compliance. Qualifying the product with a specific equipment requires a detailed evaluation of the machine parts and sterilization process to avoid contamination and batch failures. The personnel manufacturing the clinical batches is rigorously trained on the machine controls and process parameters. The manufacturing process for cGMP-compliant clinical batches is validated to produce consistent multiple batches to save time, reduce deviations, and avoid change control processes. Detailed documentation of how the method was developed and why the method evolved during various drug development stages is required for achieving regulatory approvals to be used on human participants.
-
- Stability testing of clinical supplies for establishing shelf life: Stability tests are performed to determine variations in the product quality due to environmental factors like temperature, humidity, and light, to establish shelf-life and storage conditions. If the drug is not tested for stability, it cannot be considered cGMP-compliant and will fail to achieve regulatory approval.
How does Vici achieve cGMP compliance in drug development?
Knowing when to comply with cGMP standards is very important during drug development to save time and money. Not following cGMP standards when they are needed can lead to expensive delays related to IND approval, poor quality of the investigational product, and may even adversely impact patient safety. However, drug developers must be judicious and not perform early-stage R&D under cGMP labs as it will unnecessarily slow down the project and increase cost.
At Vici, we have the expertise and knowledge to guide you achieve quality outcomes within the regulatory framework. One way Vici achieves cGMP compliance is by monitoring and tracking records, personnel training, equipment calibration, quality control, and facility design through a well-structured quality policy and Quality Management System (QMS).
Difference between clinical and commercial manufacturing
The process to manufacture drugs for clinical trials varies significantly from the process for commercial scale manufacturing due to the flexibility needed for early-stage formulation development. Even though both processes are cGMP-compliant, clinical manufacturing has its unique challenges such as non-uniform batches. Regulatory authorities may also request changes to the formulation during the Phase I and II of clinical trials. However, most issues can be solved by collaborating with the CDMO as they can streamline the drug development process and ultimately increase your chances of a successful clinical trial. Operators manufacture commercial batches of drugs in the presence of an analytical team to optimize process controls and process parameters.
Clinical Manufacturing:
-
- Since formulations are not finalized, there needs to be a certain flexibility while implementing cGMP guidelines, since it demands addressing unforeseen obstacles that may be resolved through changing the dosage.
-
- The formulation is subject to change and is often non-uniform. As clinical trials progress, the formulation is further optimized and refined.
Commercial Manufacturing:
-
- Formulations and dosages are finalized and making adjustments during this phase is generally more difficult.
-
- Drug batches are uniform ensuring quality, consistency and integrity.
Consistency and quality are the heart of Vici for drug development. While manufacturing batches for phase I and II clinical trials, Vici implements cGMP guidelines and quality control measures including, but are not limited to, pH levels, temperature, flow rates, sampling frequency, and time intervals allowing us to deliver safe, effective clinical trial materials.
Quality Standards
The Code of Federal Regulations (CFR) is the collection of general and permanent regulations from the federal government. The CFR includes the regulations enforced by the federal agencies. Title 21 CFR which belongs to the FDA explains the Federal Food, Drug, and Cosmetic Act and its implementing rules, such as the Public Health Services Act. Below are the CFRs that explain the cGMP guidelines required to ensure compliance.
-
- 21 CFR 210 – Current Good Manufacturing Practice in Manufacturing, Processing, Packing, Or Holding Of Drugs; General
-
- 21 CFR 211 – Current Good Manufacturing Practice for Finished Pharmaceuticals
References:
Varma, S. (2001). Targeting R&D quality and compliance training for technology transfer into the GMP environment of production. The Quality Assurance Journal, 5(2), 85–92. https://doi.org/10.1002/qaj.132
Deshmukh, A. S., Dighe, P. R., Mahajan, V. R., Kunde, V. D., Mhaske, G. S., & Awate, S. S. (2023). Comprehensive Analysis of Quality Management in pharmaceutical manufacturing process. Advanced Concepts in Pharmaceutical Research Vol. 2, 12–23. https://doi.org/10.9734/bpi/acpr/v2/6663e
The Center for Professional Innovation and Education. (2024, November 4). Understanding cgmp: What current good manufacturing practices are and why they matter. Pharmaceutical, biotech, medical device training courses. https://www.cfpie.com/understanding-cgmp-what-current-good-manufacturing-practices-are-and-why-they-matter
Bourgeois, A., & Paul, L. (2024, July 23). Good manufacturing practices: When do they apply?. Advarra. https://www.advarra.com/blog/good-manufacturing-practices-cgmp/
Miller, K. (2020, April 30). Deviation Management: Taking GMP Compliance to the Next Level. November 13, 2024, https://www.iqvia.com/locations/united-states/blogs/2020/04/deviation-management-taking-gmp-compliance-to-next-level/



