


Dissolution testing of pharmaceutical drug products is one of the most important tools during drug development and for quality assurance during quality control (QC) release testing. This testing is mandatory for all oral solid dose, oral semi solid dose, and oral suspension products. It is not required for drugs that are already in solution such as oral liquid dose or topical formulations.
1. What is dissolution testing?
Dissolution testing provides a measure of the rate of drug release from tablets, capsules, or liquid suspensions. During dissolution testing, the dosage form – usually solid oral dosage forms like a tablet or capsule – is placed in a stirred vessel containing a dissolution medium maintained at a constant temperature of 37 ± 0.5°C. The concentration of drug in the dissolution medium is then measured as function of time by analyzing samples withdrawn using a high-performance liquid chromatography (HPLC) machine.
Due to the importance of dissolution testing for pharmaceutical products, the United States Pharmacopeia (USP) publishes dissolution specifications for most FDA-approved products. In addition, the FDA Dissolution Method Database lists dissolution methods for products that do not have a published USP method.
2. Why is dissolution testing performed?
Dissolution testing performs two vital and separate functions during drug development and manufacturing. It is used during R&D to predict bioavailability and during manufacturing as a quality control (QC) tool.
3. How is dissolution used during drug development?
It is used during R&D to predict the drug release rate from the dosage form prior to absorption. This is the first step in understanding bioavailability. The rate of drug release from the dosage form depends on the solubility of the drug, composition and level of inactive ingredients (excipients) in the dosage form, drug substance (API) particle size, manufacturing process, and the medium surrounding the dosage form. Dissolution methods that are able to discriminate between different formulation characteristics of the same product are called discriminating dissolution methods. When determining the rate of drug release through dissolution testing, the formulation scientist can alter critical material attributes of the dosage form to optimize the formulation.
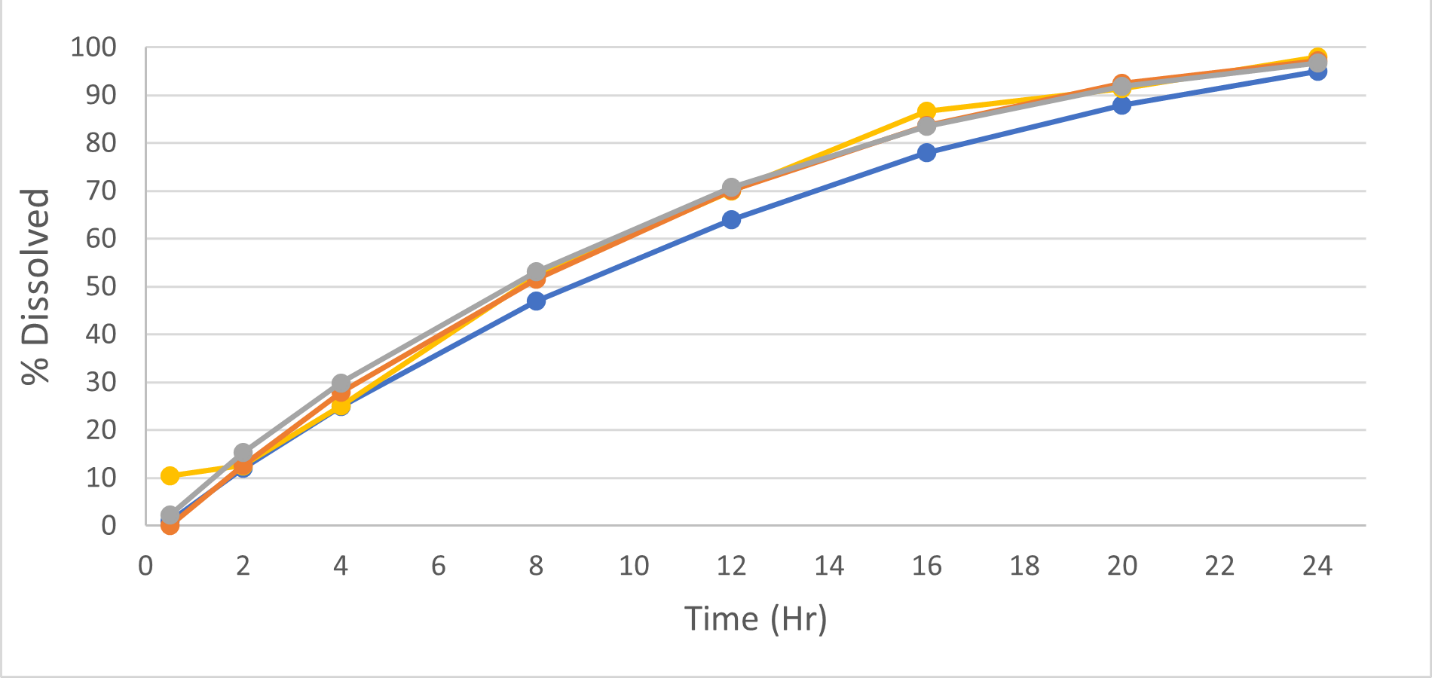
4. How is dissolution used to evaluate bioavailability and establish bioequivalence?
The drug must first be released from the dosage form prior to absorption. For low solubility drugs (BCS class II and BCS Class IV drugs), bioavailability is primarily driven by the dissolution of the drug substance. Optimizing dissolution for the formulation using discriminating and biorelevant dissolution media allows formulators to create formulations which are more likely to be absorbed in the gastrointestinal (GI) tract.
Matching dissolution profiles between two different formulations using discriminatory and biorelevant dissolution methods is likely to result in both formulations being bioequivalent. Formulation scientists frequently utilize dissolution testing to predict bioequivalence for oral solid dose formulations.
Biorelevant dissolution methods are discriminatory to different pharmacokinetic outcomes in human or animal models. They may also be used to predict dissolution of the dosage form in the presence of food to understand the food effect using simulated fasted and simulated fed dissolution medium. It is common to use Fed State Simulated Intestinal Fluid (FeSSIF) and Fasted State Simulated Intestinal Fluid (FaSSIF) during drug development.
5. Dissolution testing for generic product development and ANDA filing
Dissolution testing is critical for the development of oral solid dose generic formulations suitable for abbreviated new drug application filings (ANDA) since such products must be bioequivalent to the reference-listed drug (RLD) or reference standard (RS) product. Dissolution is performed across the pH range experienced by the dosage form to simulate its transit through the GI tract.
6. Dissolution testing as a QC tool
Since the dissolution profile can be used to discriminate between various formulation attributes of the dosage form, dissolution testing is used as a QC test during drug manufacturing and stability testing for clinical and commercial products. The FDA also views dissolution of a diagnostic tool to evaluate changes requested by manufacturers to approved products that may adversely impact bioavailability and product quality.
7. How are specifications set for dissolution testing?
Products manufactured commercially must meet dissolution specifications that are approved by the FDA during NDA or ANDA filing. For immediate release oral solid dose products, it is typically required that at least 80% of the drug is released during dissolution by a specified time, often 45 minutes. However, the time and required release percentage may vary depending on the specific details of the product. The dissolution medium, apparatus, and test method used are also specified in the approved method. For controlled release products, the rate of drug release is critical. The FDA recommends selecting at least three time points spanning the release profile for such products and typically allows a tolerance window of no more than 20% for such profiles. For enteric tablets, the dissolution test is performed at different pH conditions sequentially with specifications set during each stage.
8. What should companies do if dissolution fails specifications?
Failure to meet specifications for a commercial product result in an OOS investigation (out of specification) to understand and control the source of variation. Such OOS events may arise due to a variety of reasons and should be solved by CMC experts who have a diverse background in analytical chemistry, formulation development, material science, and manufacturing process engineers.
9. How is dissolution testing used to manage changes to commercial pharmaceutical products?
Dissolution testing and comparison is mandatory for oral solid dosage forms and oral suspension formulations when considering changes to the site, API source or grade, changes to excipient levels or source, or changes to the manufacturing process. Manufacturers must demonstrate that the changes made to approved products do not result in changes to the dissolution profile. The FDA evaluates such changes per the SUPAC-IR and SUPAC-MR framework and pharmaceutical companies must understand the impact of such changes on dissolution prior to engaging in such modifications.
10. Do dissolution methods need to be validated?
Due to the wide-ranging applications of dissolution in drug development and manufacturing, yes, the dissolution method must be developed and validated to ensure accuracy and consistency in results.
Providing testing solutions for you
Dissolution testing is a critical aspect of drug development, manufacturing, and testing. Understanding regulations and applying the available tools related to this technique requires expertise in formulation development, analytical testing, and FDA regulatory compliance. Vici Health Sciences provides comprehensive support and services for all your dissolution testing needs. Contact us to learn more about our testing solutions.



