


Quality control is an integral part of the drug development process, and the quality of analysis can make a difference in a drug product’s safety and efficacy. QC-friendly analytical methods are simple to operate and require minimum sample preparation. They are robust, reliable, and efficient for routine analytical tests. Furthermore, the analytical tests run by QC-friendly techniques are validated analytical tests which are cost-effective and easy to transfer from one laboratory to another. Unlike the exploratory methods used in R&D research, these methods are developed to yield highly reliable, consistent results across a wide range of batches and different environmental conditions. QC-friendly analytical methods are widely used to analyze various components of drugs, including active ingredients, impurities, and excipients.
Why are QC-Friendly Analytical Methods Important?
One of the major areas of focus in the importance of QC-friendly methods is the adherence to the regulatory requirements. Organizations such as the Food & Drug Administration (FDA) or European Medicines Agency (EMA) have several mandates for analytical tests that may be required to guarantee safety of pharmaceutical products. Once validated, these tests augment the stringency of quality requirements for attributes throughout the life of a product, and foster compliance and confidence in the profession.
Moreover, QC-friendly analytical approaches improve the efficiency of the drug development process. Working with Quality by Design (QbD) principles enables a broader perspective on the drug development process which allows for early detection and determination of efficient critical quality attributes (CQAs) in the early stages of QbD.
Benefits of QC-friendly Analytical Methods:

QC-Friendly Analytical Methods Development Process
Developing QC-friendly analytical methods is a systematic process which aims to ensure reliability, accuracy, and compliance of analytical results through a few key steps:
-
- Understanding the Product and Analytical Method Selection: A detailed understanding of the physicochemical characteristics of the product or analyte is required. Based on these characteristics, suitable analytical techniques such as HPLC, FTIR, NMR, and GC-MS are selected. The information generated from such techniques will help tailor the analytical method to measure CQAs like potency, purity, and stability. The analytical test must be able to assess the drug product’s potency at every point of its shelf life, even with degradation. An appropriate analytical test should be simple in execution yet calibrated enough to detect impurities and differences between each batch.
-
- Validation of Analytical Method: The selected analytical method should be validated to ensure its suitability for the intended use. Validation consists of accuracy, precision, specificity, detection limit, and linearity requirements which are specified in international guidelines, e.g. ICH Q2(R2) and USP guidelines.1,2 The next step is to tailor the parameters of the analytical test, such as HPLC conditions or GC-MS temperatures, to enhance sensitivity or reduce the time needed to complete analysis. This ensures both the reliability and efficiency of the analytical test.
-
- Continuous Monitoring and Improvement: There is a final validation that is independent of the method but is related to the working environment where the method will be, for example, implemented by trained personnel following standard operating procedures, equipment calibration, etc. In this stage, the performance of the method is consistently monitored to ensure it remains feasible, addresses needs, and can be optimized when necessary. This way, the method does not become obsolete and can satisfy regulatory requirements wherever applicable.
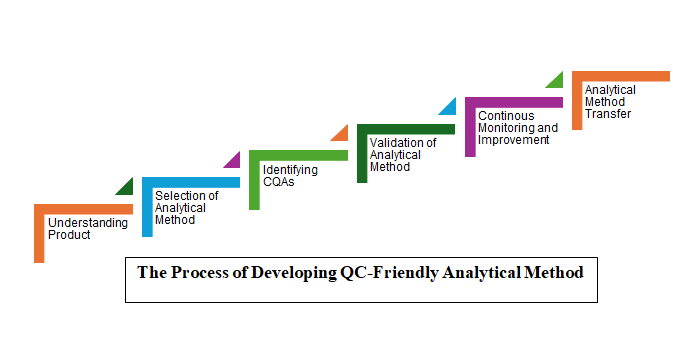
Analytical Method Transfer
Analytical method transfers (AMT) are one of the important steps of drug development and can often be challenging to do so. Transferring a method within the same company can sometimes pose problems in sample stability, documentation, clarity of procedures, or underestimation of the complexity of the process. An unsuccessful method transfer can lead to out-of-specification results, further investigations, rework, costly delays, and consequences from regulatory bodies.
The real challenge for analytical methods transferred from R&D labs to QC labs, or from one lab to another, is being able to maintain their reproducibility and reliability in analytical tests. Different approaches to analytical method transfer are taken during the drug development stage, which varies as per the nature of the method. As development progresses, the requirements for successful transfer become more stringent. According to the USP General Chapter <1224>3, there are several approaches to AMT:
-
- Direct Transfer: Entails practical elaboration of the method through a visit by the sending unit (SU) to the receiving unit (RU). After all samples are collected, including those from the RU, both units/laboratories conduct sample analyses.
- Comparative Testing: Most frequently used methodology. This provides both laboratories with the same batch and a homogeneous material to test. This approach assures consistency in results with regard to a particular sample obtained from two different environments.
- Co-validation Between Laboratories: Two laboratories cooperate in the method validation. Each participate in the process of inter-laboratory validation and perform the same degree of validation.
- Revalidation: Whenever equipment, reagents, or the processes of manufacturing are modified, revalidation is essential to verify the performance of the method. This guarantees that the method is still applicable on different instruments and/or after revisions made to the applied parameters.
- Transfer Waiver: On occasions, a formal method transfer may become redundant if the RU already has the competencies to utilize the method without making comparisons or inter-laboratory data. However, this must be well documented.
Considerations for Successful Method Transfer
Even with careful planning, method transfers can encounter challenges. Here are some tips on how you can avoid common pitfalls:
-
- Proper Training: Ensure all transfer personnel are trained and have full competency on the methodology. Provide detailed standard operating procedures, references, validation reports, and any other documents that are necessary for running the method successfully. Receiving personnel should run the analytical method to identify any issues during the training.
- Simple and Reproducible Methods: The method should be simple to run and user-friendly. Documentation should include detailed explanations of objectives, scope, materials, instruments, and acceptance criteria. This provides clarity in the transfer process by all the parties involved.
- QC-Centric Approach: The SU should consider potential challenges QC scientists may face. Avoid using materials that are inconsistent, difficult to procure, or otherwise problematic to work with. Optimize sample preparation and data acquisition as much as possible. Ensure the method is flexible and can easily be adapted to different instruments.
- Effective Communication: Sending/Transferring and receiving labs must have open, consistent communication. Establishing effective communication early can help eliminate misunderstandings and ensure the proper use of methods in the QC environments.
- Proactive Risk Management: All possible risks facing the transfer shall be analyzed before the transfer. The transfer will consider method complexity, product specifications, and the capabilities of the receiving laboratory. The more thorough this analysis is, the more seamless the strategy will be during transfer.
Quality is our Standard
In the pharmaceutical industry, achieving proper product quality, regulatory compliance, and operational efficiency is critical in developing QC-friendly analytical methods. Robust, reproducible, and simple methods intended to measure the critical quality attributes throughout a product’s lifecycle can help empower its stakeholders. Analytical method transfer can ensure that there will be similar testing results between R&D environments and QC environments at scale-up; hence, specific best practices such as training, simplification, and control of risks aid in overcoming some obstacles. Developing such methods early in the drug development process minimizes the product failure risks, delays, or recalls later on.
As a company founded and led by formulation scientists, Vici has a specialized understanding of drug development and what’s required during each step of the process. Get in touch with us to learn more about how Vici upholds the highest quality standards throughout all of our client’s drug development programs.
References
1. USP. The United States Pharmacopeia (2024) Validation of Compendial Methods. 48th Edition (USP-NF 2024), Rockville. 2024.
2. ICH. International Conference of Harmonization: ICH Q2(R2) Validation of analytical procedures – Scientific guideline. 2024.
3. USP. The United States Pharmacopeia (2024) Transfer of Analytical Procedures. 48th Edition (USP-NF 2024), Rockville.2024.



