


Well over 80% of medicine prescribed in the US by volume are generics. Generic substitution saves the US healthcare sector billions of dollars and makes many medicines affordable to patients. Per the US FDA, even drugs that contain the same active ingredients at the same strength utilizing the same route of administration are not considered interchangeable unless they are bioequivalent. Demonstrating bioequivalence (BE) is a fundamental aspect of seeking and receiving generic drug approval through the Abbreviated New Drug Application (ANDA) filing pathway.
In 1977, the US Food and Drug Administration (FDA) introduced bioequivalence studies and since then has evolved it as a specific regulatory requirement for generic drug approval.1 In this article, we will explore the concept of bioequivalence and provide a step-by-step guide for evaluating bioequivalence, using immediate-release dosage forms as a case example.
What is Bioavailability?
When a drug is administered, an adequate amount of the administered dose must be absorbed over a specific amount of time time for the intended drug effect. Bioavailability is “the rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of action.” The bioavailability of an oral dosage form relies on the drug’s physical and chemical characteristics and the physiological conditions of the gastrointestinal tract. Bioavailability is an indirect measurement of how quickly and extensively the drug is released from the medication product and delivered to the site of action.
Bioavailability can be classified into:
- Absolute Bioavailability: Compares the bioavailability of an orally administered drug with the same drug given intravenously (100% absorption).
- Relative Bioavailability: Compares different formulations or routes of administration, such as comparing a new oral dosage form with a reference product.
What is Bioequivalence?
Bioequivalence is “the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administered at the same dose under similar conditions in an appropriately designed study.”
Pharmaceutically equivalent drugs have the same active ingredient, potency, dosage form, route of administration, and same label. However, these pharmaceutical equivalent products can only be labeled as bioequivalent unless they are also therapeutically equivalent.
Bioequivalence must be established for all oral solid dosage forms, oral semi solid dosage forms, and topical semi solid dosage forms. For oral dosage forms, this is done by means of a crossover human bioequivalence clinical study where the drug concentrations are measured in plasma. However, establishing bioequivalence semi solid dosage forms is more complicated because there may not be sufficient systemic circulation of the drug molecule.
Bioequivalence studies are utilized in a variety of contexts, most notably when a sponsor proposes producing a generic version of an approved novel medicine (through an ANDA). For a generic drug to be approved by a regulatory authority, it must be bioequivalent to the reference drug. Below is a simulated example of the area under the curve (AUC) of two drug products and their maximum concentration (Cmax) versus time (Adapted from Faull 2003) .2
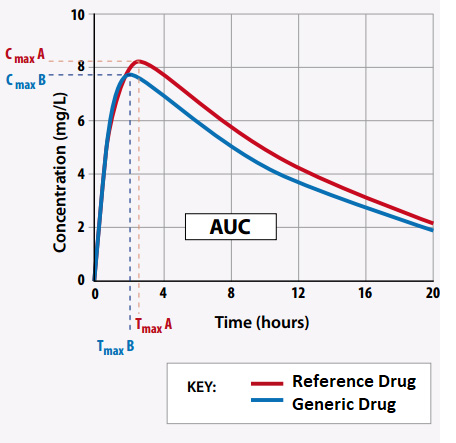
Reference Drug: CmaxA = 8.1 mg/L; Tmax = 2.6 h; AUC = 124.9 mg.h/L
Generic Drug: CmaxB = 7.8 mg/L; Tmax = 2.5 h; AUC = 120.1 mg.h/L
To be considered bioequivalent, the area ratio’s 90% confidence intervals must lie between 0.8 and 1.25. In this scenario the generic drug to brand drug ratio is 0.96 and can be considered bioequivalent.
Process for Conducting Bioequivalence Studies
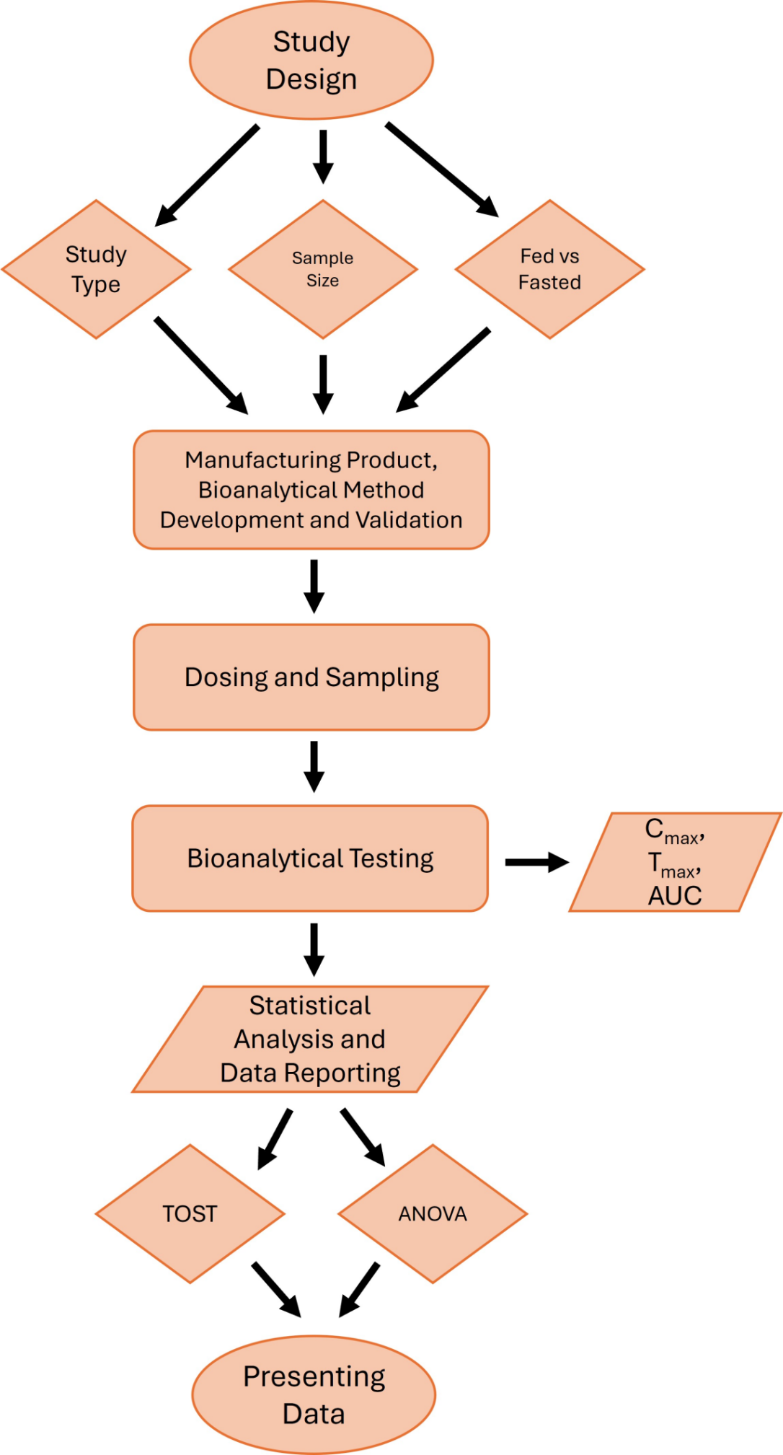
Step 1: Study Design
Generally, crossover designs are used for analyzing pharmacokinetic parameters to evaluate the bioequivalence between the generic and reference drug. The benefit in crossover designs is that the same subject is exposed to the test and reference drug sequentially. This reduces variability intrasubject variability when comparing the test and reference for that individual. Such bioequivalence studies are also performed in both fed and fasted state because of possible food effects. Food effect is said to be observed when meals influence the rate and extent of drug absorption.
Pharmacokinetic Study Design
- Study type: Single-dose, crossover study
- Design: The standard design is a two-period, two-treatment, two-sequence randomized crossover design. In this study design, both generic and reference brand name drugs are administered to each subject, with a washout phase in between to avoid carryover effects. Typically, in this type of study design, the subjects are split randomly into two groups. One group receives the test product first and then the reference products and the order is switched for the second group.
- Sample size: The sample size is based on the expected variability in pharmacokinetic profiles for the drug molecule and is often determined through literature review for generic drugs. Regulatory guidelines specify that there should be a statistical justification for the number of subjects but must not be fewer than 12. Allowances must be made for dropouts while determining the number of subjects needed.
- Washout period: A sufficient washout period should be included to ensure no drug remains in the system before the second administration (usually dependent on the drug’s half-life).
Step 2: Manufacturing, bioanalytical method development, and validation
Manufacturing Product for Bioequivalence Studies
Key steps required during ANDA drug development prior to manufacturing the test product are:
-
- Formulation Development: Developing a formulation that matches the reference product in key attributes such as dissolution rate, solubility, and particle size.
- Analytical Method Development: Developing analytical methods to determine the drug dose and rate of release and compare this with that of the reference listed drug (RLD) or reference standard (RS).
- Process Development: Establishing a reproducible manufacturing process which meets regulatory requirements and ensures consistency across different batches.
- Quality Control: Implementing rigorous quality control measures to monitor the stability, purity, and potency of the product and ensure the product meets predefined specifications.
- GMP Compliance: Manufacturing should follow Good Manufacturing Practices (GMP), ensuring safety and compliance with regulatory standards.
Bioanalytical Method Development
Bioanalytical methods are developed to measure the active drug and its metabolites in biological matrices (e.g., plasma or serum) for the determine pharmacokinetic parameters. This is different from analytical method development testing for the test product in that bioanalytical methods are specifically developed to measure drug in biological fluids. It is important to understand that these methods are not interchangeable. Typical techniques for bioanalytical assays include liquid chromatography-mass spectrometry (LC-MS) for small molecule or peptide drugs and enzyme-linked immunosorbent assays (ELISA) for biologics. The bioanalytical test method must be able to measure both unbound and protein-bound drug in blood or plasma.
Method Validation
Bioanalytical methods must be validated according to regulatory guidelines (e.g., FDA, EMA) to ensure they are accurate, precise, specific, and reliable. Key validation parameters include accuracy, specificity, linearity, limit of detection (LOD) and limit of quantitation (LOQ), and sample stability
By comparing the pharmacokinetic parameters (e.g., C-max, AUC) of the test product to the reference, regulatory authorities can assess bioequivalence. Ensuring the accuracy and reliability of bioanalytical methods is crucial for these assessments to be valid and acceptable in the approval process.
Step 3: Dosing and Sampling
Proper dosing and sampling is, naturally, very important for accurate results. Parameters such as dose selection, drug administration conditions and sampling frequency play a critical role during the determination of the BE studies. Key parameters that should be considered during BE studies include:
Dosing in Bioequivalence Studies
Single vs. Multiple Dose:
Single Dose Studies:
-
-
-
- Generally performed for drugs with linear pharmacokinetics.
- A single dose is administered under controlled conditions (fasting or fed, as required).
-
-
Multiple Dose Studies:
-
-
-
- Required for drugs with nonlinear pharmacokinetics or long half-lives.
- Study conducted after achieving steady state.
-
-
Dose Selection:
-
- The dose used should be clinically relevant.
- The highest marketed dose is usually tested to maximize sensitivity to differences in bioavailability.
Administration Conditions:
-
- Fasting state: Standard, unless the label specifies administration with food.
- Fed state: Required for drugs with significant food effects or as per regulatory guidelines.
- The same molar dose for both generic and reference brand name drugs is administered.
Sampling in Bioequivalence Studies
- Plasma or Serum Sampling:
-
-
- Blood samples are collected at specific intervals to capture the drug’s pharmacokinetic profile.
- Frequency and Duration:
-
-
- Frequency: Frequent sampling is required around Tmax to accurately capture Cmax.
- Duration: Sufficient sampling post-dose to cover at least 80–90% of the AUC (typically up to 5–7 half-lives).
- Number of Samples:
-
-
- Generally, 12–18 samples per subject are adequate.
- The precise number depends on the drug’s half-life and the study’s design.
Step 4: Bioanalytical Testing
After drug administration, blood samples are collected at various time intervals to determine the pharmacokinetic profile of the administered drugs. Parameters include:
-
- Cmax: Maximum plasma concentration of the drug.
-
- Tmax: Time taken to reach maximum plasma concentration.
-
- AUC: Area under the curve .
-
- The elimination phase to determine half-life.
Step 5: Statistical Analysis
The statistical analysis of pharmacokinetic parameters in BE studies, as outlined in USP <1090> and FDA guidelines, focuses on comparing the rate and extent of drug absorption between a generic drug and a reference brand name drug.3,4
Pharmacokinetic data is log-transformed to normalize distribution before statistical analysis. Here are some common statistical approaches for BE analysis:
1. Two One-Sided Tests Procedure (TOST):
-
- The TOST procedure is a widely used method to test bioequivalence. It is designed to check if:
- The generic product is not significantly less than the brand name product.The brand name product is not significantly less than the generic product.
- The 90% confidence interval (CI) for the ratio of the geometric means of the generic and reference products is calculated for both AUC and Cmax.
- The acceptance range for bioequivalence is set between 80% and 125%. For a product to be considered bioequivalent, the 90% CI for both AUC and Cmax must fall within this range.
- The TOST procedure is a widely used method to test bioequivalence. It is designed to check if:
2. Analysis of Variance (ANOVA):
For a two-treatment, two-period, two-sequence crossover design, the ANOVA model includes the following factors:
-
- Sequence (Group/Order): Represents the order in which subjects receive the generic and the reference brand name drug.
-
- Subjects Nested in Sequence: Accounts for variability between subjects within each sequence.
-
- Period (Phase): Captures time-related differences in drug absorption between the two periods.
-
- Treatment (Drug/Formulation): Reflects differences between the generic and brand name drug formulations.
The sequence effect is evaluated with the mean square for [subjects (sequence)] as the error term, whereas other major effects are tested with the residual error mean square from the ANOVA.
Step 6: Presenting Data
When submitting BE study results, the following should be included:
- Individual concentration-time data for each subject.
- Pharmacokinetic parameters such as AUC and Cmax for both generic and reference brand name products.
- Statistical analyses of pharmacokinetic data, including 90% confidence intervals for generic drug/reference drug ratios.
Generic to Market
Bioequivalence studies in generic drug approval require detailed planning, evaluation of pharmacokinetic parameters, and statistical analyses to determine therapeutic equivalence between generic and reference drug products.
It is important to have a clear understanding of bioequivalence and why it matters for ANDA drug development. At Vici, we understand the science and regulatory aspects of why and how to go about conducting BE studies. Contact us to learn more about how Vici can help you accelerate your drug product to market.
References
1. Chen M-L, Shah V, Patnaik R, et al. Bioavailability and bioequivalence: an FDA regulatory overview. Pharmaceutical research. 2001;18:1645-1650.
2. Faull R, Hendry MM, Berkovic SF, Vajda FJE, Birkett D. Generics-equal or not? Australian Prescriber. 2003;26(6):85.
3. Guidance FDA. Statistical approaches to establishing bioequivalence. Center for Drug Evaluation and Research. 2001;
4. USP. The United States Pharmacopeia (1090〉 Assessment of Drug Product Performance. 48th Edition (USP-NF 2024), Rockville. 2024;



